A research team has recently found that a common antifungal drug may be an effective treatment for patients with cystic fibrosis. These results come from a study in which human cells and animal models of the disease were found to respond to the medication, amphotericin. When administered the drug, the lung cells began functioning in a manner that could help patients fight the chronic bacterial lung infections commonly associated with cystic fibrosis. Carried out by researchers from the University of Illinois and the University of Iowa and supported in part by the National Heart, Lung, and Blood Institute (NHLBI) part of the National Institutes of Health, this work will appear in the journal Nature.
“Instead of trying do gene therapy – which is not yet effective in the lung – or to correct the protein, our approach is different. We use a small molecule surrogate that can perform the channel function of the missing protein, which we call a molecular prosthetic,” said Dr. Martin D. Burke, study leader, professor of chemistry at Illinois, and associate dean for research at the Carle Illinois College of Medicine.
Cystic fibrosis is a disease that persists throughout one’s life and increases susceptibility to lung infections. Treatment options exist for some patients; however, no cure has been identified. Use of amphotericin restored immune function of lung tissue in human samples as well as in pigs with cystic fibrosis. The drug worked effectively regardless of the genetic mutation causing the disease, potentially making it the first treatment that can apply to all types of cystic fibrosis.
READ MORE: New Treatment Affective Against Drug-resistant Bacteria
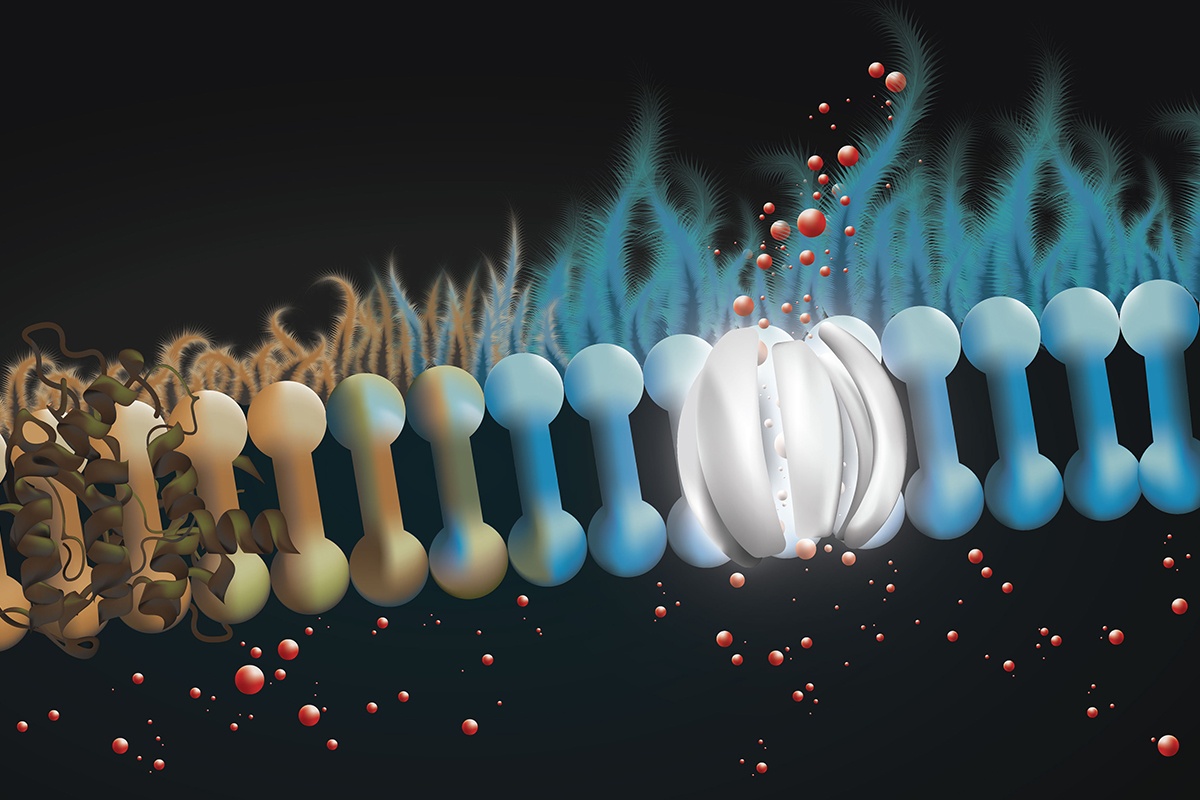
The layer of liquid coating the surface of the lungs help protect against infection, but excessive thickening of this mucus layer is often associated with the disease. Cells that line the lung secrete bicarbonate into the liquid layer to regulate pH and make it uninhabitable to infectious bacteria. Cystic fibrosis is characterized by the lack of functioning GFTR, a membrane protein needed to channel bicarbonate to the surface.
New Approach to Treatment
Burke’s group has been researching the channel-forming properties of amphotericin for years. In this newest study, the group aimed to use the medication in treating cystic fibrosis. The team found that amphotericin was able to form channels on the surface of lung tissue membrane’s donated by people with cystic fibrosis caused by mutation in the CFTR gene. These channels released bicarbonate that had been built up in cells and reduced the pH and thickness of the liquid on the lung membrane back to a normal range.
Unlike medications that target the defective CFTR proteins and work to adjust the improper protein folding, the use of amphotericin bypasses the defected protein by forming new channels. Also, in the 10% of cystic fibrosis patients who lack the protein altogether, there is no option to correct CFTR structure being that it is absent. This new approach to treating cystic fibrosis is especially profound in these cases, where it may be the only option.
“Whereas many of the latest advances in cystic fibrosis treatment have been targeted to specific mutations, this approach would benefit everyone with cystic fibrosis, regardless of mutation,” said Emily Kramer-Golinkoff, co-founder of the nonprofit cystic fibrosis research foundation Emily’s Entourage, which partially funded the research. “Second, and perhaps even more importantly, this approach presents an opportunity to repurpose an existing, approved drug and bring it to the clinic quickly.”
READ MORE: Cystic Fibrosis and Rare Inflammatory Diseases
Going forward, the Illinois-Iowa research team intends to conduct clinical trials to test amphotericin’s efficacy in treating humans with cystic fibrosis.
“Since amphotericin is an already approved drug, the path to clinical translation is more direct. It’s already been shown to be safe when delivered directly to the lung, and it doesn’t get into the rest of the body, so we can avoid the negative side effects that the drug is known for,” said Burke. “We are hoping to conduct clinical trials soon, especially with people for whom correctors are not beneficial.”
“The cystic fibrosis community is truly in need of new therapies to reduce the burden of this disease. We are interested to see how this potential treatment performs in clinical trials in the future,” said Dr. James Kiley, director of the Division of Lung Diseases at NHLBI.
Exciting research from a team led by @ILLINOISmed faculty Dr. Marty Burke, published in @nature: An approved drug normally used to treat fungal infections has potential to become the first treatment to address all types of cystic fibrosis.https://t.co/Cx4HpWZsCv #ILLINOIS
— University of Illinois Research Park (@UIResearchPark) March 14, 2019
READ MORE: FDA Approves Ketamine-Derived Nasal Spray Treatment for Depression
Source: University of Illinois





 © 2025 Mashup Media, LLC, a Formedics Property. All Rights Reserved.
© 2025 Mashup Media, LLC, a Formedics Property. All Rights Reserved.